




BIOINFORMÁTICA PRÁTICA EM MODELAGEM MOLECULAR
Criando uma caixa d’água
Crie uma pasta com nome “Caixa d’água”. Dentro da pasta, abra o terminal do Linux. Isso pode ser feito clicando com o botão auxiliar na tela e selecionando a opção Abrir o Terminal Aqui. Digite vmd e dê enter para abrir o programa VMD. Na barra de tarefas, clique em Extensions, posicione o mouse sobre a guia Modeling e clique em Add Solvation Box.

Surgirá uma nova janela. Em Output estará escrito solvate, esse será o nome dos arquivos que serão gerados no processo. Vamos alterá-lo para agua .
Box Size e Box Padding apresentam espaços para preenchimento das coordenadas de nossa caixa d’água. Criaremos uma caixa de 16Å (Angstroms) no mínimo e 16Å no máximo em todas as direções, x, y e z.

Isso gerará um cubo com aresta de 32Å . Você pode assim calcular o volume de nossa caixa d’água com um calculo simples:
V= a³, assim V da caixa d’agua= (33Å) ³ = 32 768Å ³ 1 milimetro = 10 000 000 Å
Para se ter uma idéia, uma gota de água tem um volume de aproximadamente 50mm³ (0,05mL), que equivalem por sua vez à 500 000 000ų. É como se pegássemos uma gota d’água e a dividíssemos quase 15000 vezes! Ou seja, estamos falando de uma quantidade extremamente pequena de água, impossível de se perceber à olho nu. Clique em Solvate. Surgirá na janela de visualização sua caixa d’água e serão gerados arquivos psf, pdb e log com o nome agua, conforme o output que escolhemos.

Feche o programa. Isso pode ser feito clicando em File na barra de tarefas e em seguida em Quit.
Para gerar uma simulação precisaremos também de um arquivo de configuração que dirá basicamente sob quais condições a simulação deverá ser gerada, como por exemplo, sua temperatura e o tempo da simulação. Esses arquivos podem ser são oferecidos para download gratuito pela internet e também são fornecidos pelo e-mail: bioinformaticapratica@gmail.com .
São arquivos do tipo conf. Será necessário editar o arquivo para que ele seja utilizado para gerar a simulação. Também precisaremos de um arquivo de parâmetros para o campo de força. É uma arquivo do tipo inp. Utilizaremos o campo de força utilizado para simulações com lipídios e proteínas, que será o mesmo utilizado para as próximas simulações, para efeito de comparação. Vamos explorar e editar nosso arquivo de configuração. Clique com o botão auxiliar sobre o arquivo e clique em “Abrir com o KWrite”. Surgirá uma janela de texto conforme a figura abaixo.
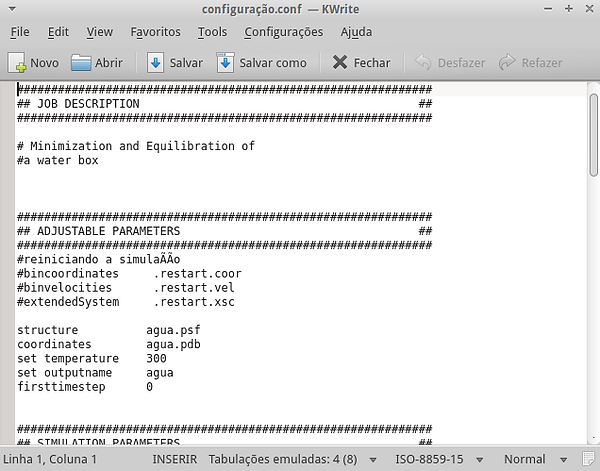
Sempre que encontrar # no inínio de uma linha, significa que aquela linha não será lida pelo programa, mas esses dados são importantes para quem acessar o arquivo saber do que se trata. A primeira linha contém o nome da nossa simulação em inglês que será “Minimization and Equilibration of a water box”. As próximas informações, que estão em destaque na figura dizem a partir de quais arquivos a simulação será gerada, sendo eles os arquivos psf e pdb que geramos com o VMD; o output, que é o nome dos arquivos de saída, ou seja, o nome dos arquivos que serão gerados pela simulação; a temperatura em Kelvin; o ponto a partir do qual simulação começará. Essas informações deverão estar da seguinte maneira:
structure agua.psf
coordinates agua.pdb
set temperature 300
set outputname agua
firsttimestep 0
Rolando para mais à baixo no arquivo, preencheremos os dados do arquivo de parâmetros, que será o arquivo par_all27_prot_lipid.inp, fornecido também pelo nosso e-mail: bioinformaticapratica@gmail.com e confirmaremos a temperatura desejada com o comando $temperature.
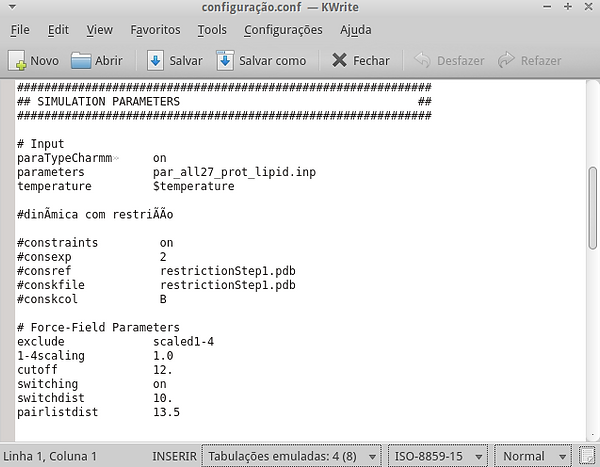
Esse campo deve estar da seguinte forma:
paraTypeCharmm on
parameters par_all27_prot_lipid.inp
temperature $temperature
O próximo campo que iremos preencher diz respeito às coordenadas da caixa d’água. Para saber essas coordenadas, precisaremos abrir os arquivos psf e pdb gerados com o VMD. Para tanto, abra o terminal na pasta onde estão os arquivos, digite vmd. Na barra de tarefas do VMD, clique em File e escolha a opção New Molecule.

Surgirá uma nova janela. Clique em Browse, selecione agua.psf e clique em Load. A janela permanecerá aberta, repita o processo dessa vez selecionando o arquivo agua.pdb.

Feche essa janela. Será possível ver sua caixa dágua na janela de visualização. Na barra de tarefas, clique em Extensions e escolha a opção TkConsole. Surgirá o terminal do VMD.
Digite os seguintes comandos, pressionando enter para cada linha para que o comando seja aceito:
set all [atomselect top all]
measure minmax $all

Surgirá as coordenadas de x, y e z, conforme destacado na imagem. O último X deve ser subtraído do primeiro para achar sua coordenada. o mesmo deve ser feito para Y e Z. Essas coordenadas devem ser editadas no arquivo de configuração, sempre usando números arredondados para mais. Da seguinte forma:
cellBasisVector1 32. 0. 0.
cellBasisVector2 0. 32. 0.
cellBasisVector3 0. 0. 32.
Sendo 32 o valor arredondado para mais fruto da subtração das coordenadas fornecidas pelo VMD.
Precisamos ainda das coordenadas do centro. Para tanto, ainda no terminal do VMD, digite:
measure center $all
Surgirão as coordenadas do centro.

Copie e cole essas coordenadas na seguinte linha do arquivo de configuração:
cellOrigin 15.982138633728027 15.962928771972656 16.041030883789063
Sendo os números as coordenadas do centro fornecidas pelo VMD.
Precisamos ainda editar o campo PME. Para tanto, devemos usar números que sejam múltiplos de 2, 3 ou 6 de forma que se aproxime do valor de nossas coordenadas, sempre para mais. Nesse caso, como nossas coordenadas são todas 32, podemos usar o próprio 32 que é múltiplo de 2. Você deverá obter algo assim:
PME yes
PMEGridSizeX 32
PMEGridSizeY 32
PMEGridSizeZ 32

Por último editaremos o tempo da simulação, que será nesse caso 200000 fs, equivalendo à 2 x 10^-10 s ou 400 ps.
run 200000 ; #400 os
Está pronto nosso arquivo de configuração.
Agora que temos todos os arquivos necessários, psf, pdb, conf e inp, em nossa pasta, podemos usar o NAMD para gerar a simulação. Para tanto, na pasta, abra o terminal e digite:
namd2 configuração.conf > configuração.log &
Esse comando irá gerar a simulação e um arquivo tipo log com o nome configuração que dirá caso algo saia errado. O uso de & permite que você continue a usar o terminal para outras atividades.
Obs: Levará algum tempo até gerar a simulação variando de acordo com o computador usado.
Vários arquivos serão criados durante o processo. Para visualizar a simulação, abra o VMD na pasta. Com o VMD, abra os arquivos água.psf e água.dcd.
Assim, nossa simulação está completa.