




BIOINFORMÁTICA PRÁTICA EM MODELAGEM MOLECULAR
Comportamento de um hidrocarboneto em água
Crie uma nova pasta com o nome hexano. Insira nessa pasta uma cópia dos arquivos configuração.conf, geral.inp, top_all36_cgenff.rtf, geral.pgn e par_all27_prot_lipid.inp. Para criar nosso hidrocarboneto será preciso primeiro criar nossos arquivos pdb e psf. Para tanto vamos lançar mão do Avogadro, editor molecular. Abra o Avogadro e na barra de tarefas, clique em Build, Insert, Fragment. Surgirá uma nova janela.

Clique duas vezes sobre a pasta alkanes, selecione a opção hexane e clique em Insert. Surgirá na tela uma molécula de hexano. Salve o arquivo, clicando em Save As. Na nova janela nomeie o arquivo, selecione o tipo pdb. Certifique-se que o arquivo está sendo salvo na pasta que foi criada.

Feche o programa. Temos nosso arquivo pdb. Antes de criar o arquivo psf, vamos aumentar o número de moléculas de hexano no arquivo pdb. Para isso, lançaremos mão de um novo programa, o Packmol. Também precisaremos de um arquivo tipo inp. Que deverá ser editado. Esse arquivo é fornecido junto desta apostila. Abra seu arquivo .inp usando o KWrite.
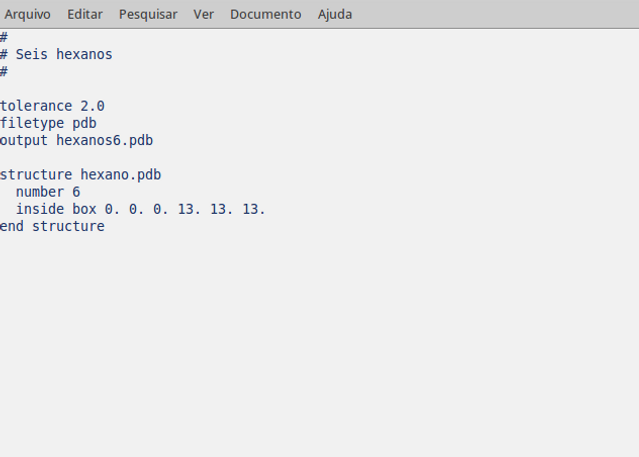
As linhas que deverão ser editadas são:
1) O título do arquivo, que não é lido pelo programa:
#
# Seis Hexanos
#
Nesse caso, escolhemos o nome Seis Hexanos porque corresponde o número de moléculas de hexano que desejamos no arquivo final, mas poderia ser quantas moléculas você desejasse.
2) A tolerância, o tipo de arquivo que desejamos gerar e o nome no arquivo gerado:
Tolerance 2.0
Filetype pdb
Output hexanos6.pdb
3) A estrutura do qual o programa deve partir, que será aquela que criamos no Avogadro; o número de moléculas que desejamos ter no arquivo final; o tamanho da caixa, ou seja, a distribuição dessas moléculas no espaço, que nesse caso escolhemos 13Å:
Structure hexano.pdb
number 6
inside box 0. 0. 0. 13. 13. 13
end structure
Como desejamos partir do eixo, as três primeiras coordenadas foram 0.
O tamanho de 13Å foi escolhido em função da distância das ligações entre carbono e hidrogênio, para que uma hexano ao ser colocado ao lado de outro não sofra sobreposição.
Está pronto nosso arquivo de input.
Agora é só salvar o arquivo com o nome hexanos6.inp e fechar. Abra o terminal na pasta. Certifique-se que esta pasta contenha os arquivos .pdb e .inp criados. Digite os comandos: Packmol < hexanos6.inp
Será gerado um arquivo hexanos6.pdb conforme o output que escolhemos.
Abra esse arquivo no VMD para visualizá-lo se desejar.
Agora vamos criar um arquivo psf. Para tanto precisaremos editar nosso arquivo .pdb para que ele seja reconhecido. Abra esse arquivo usando o KWrite.
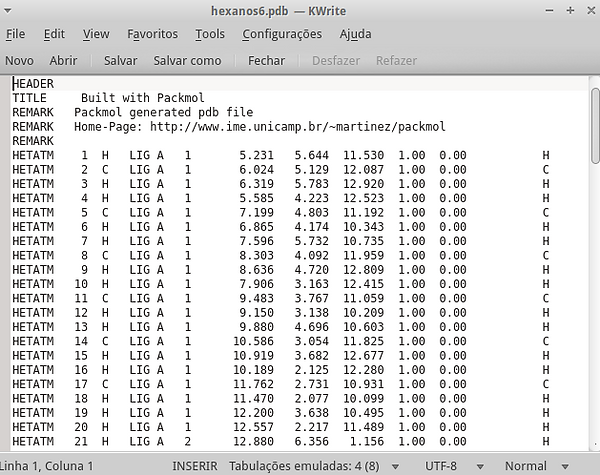
Note que na terceira coluna você encontra o nome de qual átomo que se trata, se é carbono ou hidrogênio, mas não estão enumerados. Para numerá-los precisaremos fazer uso do arquivo de topologia (top_all36_cgenff.rtf.) fornecido em nosso e-mail: bioinformaticapratica@gmail.com. Abra esse arquivo usando o KWrite. Abra o localizador (isso pode ser feito digitando Ctrl+f) e digite hexane para procurar a estrutura do hexano.

Esse arquivo te fornece não só a identificação de cada átomo, mas também o nome do resíduo. Você deverá editar linha por linha. Na coluna GROUP você encontra a identificação no átomo.
ATENÇÃO: Cada dígito que você aumentar na linha ao acrescentar o número do átomo, você deverá deletar de espaço vazio à frente, de forma que a linha editada permaneça do mesmo tamanho da linha abaixo que ainda não sofreu edição.
O nome do resíduo também deverá ser substituído, de LIG para HEXA conforme o arquivo de topologia.
Como a molécula de hexano se repete, a molécula 2 receberá os mesmos nomes da molécula 1, assim como a 3, a 4, a 5 e a 6. Ao final o arquivo pdb deverá estar como na imagem abaixo.
Dica: como o nome do resíduo se repete em todas as linhas, você pode selecionar a coluna e fazer a substituição de uma só vez. Para isso digite Ctrl+Shift+b, selecione a linha LIG, delete e digite HEXA. A linha inteira será substituída!
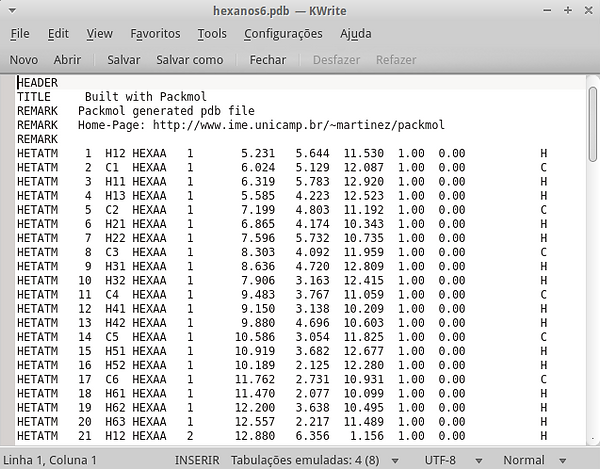
Salve e feche o arquivo.
Para gerar o .psf precisaremos de um arquivo .pgn fornecido no email: bioinformaticapratica@gmail.com
Esse arquivo deve ser editado. Nesse arquivo deve constar o arquivo de topologia que será usado, o arquivo pdb do qual o programa partirá e o nome dos arquivos de psf e pdb que serão gerados.
O arquivo de topologia:
topology top_all36_cgenff.rtf
O arquivo de pdb, que será aquele que geramos através do Packmol:
Segment A { pdb hexanos6.pdb }
coordpdb hexanos6.pdb A
O nome dos arquivos gerados:
writepdb Hexanos6.pdb
writepsf Hexanos6.psf
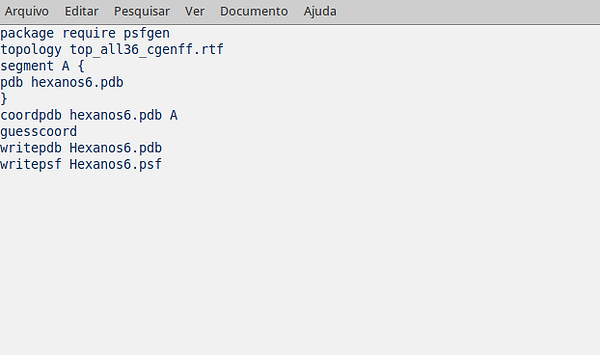
Salve o arquivo com nome hexanos6.pgn e feche o arquivo. Deverão estar na pasta os seguintes arquivos: hexanos6.pdb
top_all36_cgenff.rtf
hexanos6.pgn
Abra o terminal nessa pasta e digite:
vmd –dispdev text –e hexanos6.pgn
Serão gerados os arquivos Hexanos6.pdb e Hexanos6.psf.
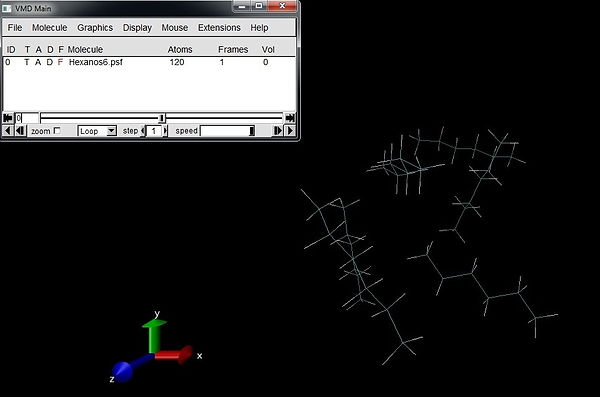
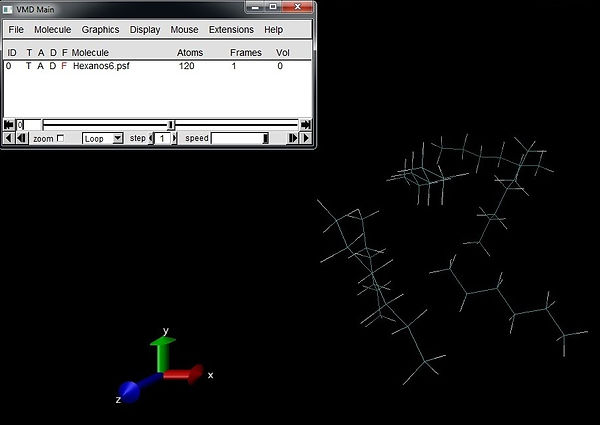
Abra os arquivos com o VMD. Surgirá na tela as moléculas de hexano.
Vamos agora solvatá-las.
Na barra de tarefas, clique em Extensions, escolha a guia Modeling e clique em Add Solvation Box.
Você perceberá que os arquivos psf e pdb já estarão pré-carregados na nova janela.
Altere o nome do output para solvatehexanos6 na caixa destacada em amarelo na figura a baixo, esse será o nome dos arquivos psf e pdb que serão gerados.
Mantenha a opção Use Molecule Dimensions (em vermelho) marcada. Em Box Padding (em azul) preencha todas as coordenadas com 12Å, esse valor possibilitará criar uma caixa d’água semelhante ao tamanho da caixa usada na solução salina e ao mesmo tempo dará uma mobilidade mais adequada às moléculas de hexano.
Clique em Solvate (em verde). As moléculas de hexano serão envolvidas por uma caixa d’água.
Feche essa janela.

Para melhorar a visualização, em Graphics, na barra de tarefas, clique em Representation.
Na nova janela, para selecionar os resíduos de hexano, digite na aba Selected Atoms a palavra lipids, pressione enter e em Drawing Method altere sua configuração para VDW. Se desejar, também poderá mudar a configuração da água por digitar water na aba Selected Atoms, em seguida clique em Create Rep e selecionar a nova configuração em Drawing Method, conforme suas preferências.
Obs: Essa etapa é apenas para facilitar a visualização e pode ser ignorada, não alterando o curso da simulação.
Feche o programa. Já terão sido criados os arquivos pdb e psf da solução.
Agora vamos editar nosso arquivo de configuração. Lembrando que esse arquivo é fornecido em nosso email: bioinformaticapratica@gmail.com.
Arquivos:
structure solvatehexanos6.psf
coordinates solvatehexanos6.pdb
set temperature 300
set outputname solvatehexanos6
firsttimestep 0
Parâmetros:
paraTypeCharmm on
parameters par_all27_prot_lipid.inp
parameters par_all36_cgenff.prm
temperature $temperature
Atenção! Não são as mesmas coordenadas demonstradas! Verifique essas coordenadas no VMD, usando o TkConsole, como demonstrado na primeira simulação:
cellBasisVector1 32. 0. 0.
cellBasisVector2 0. 32. 0.
cellBasisVector3 0. 0. 32.
cellOrigin 15.982138633728027 15.962928771972656 16.041030883789063
O mesmo vale para o PME que deve ser recalculado conforme as dimensões da sua nova caixa d’água. PME yes
PMEGridSizeX 32
PMEGridSizeY 32
PMEGridSizeZ 32
Tempo:
run 10000000 ; #20 ns
Não se esqueça de salvar o arquivo de configuração.
Agora podemos gerar a simulação com o NAMD. Abra o terminal na pasta, digite os seguintes comandos:
namd2 configuração.conf > configuração.log &
Levará algum tempo até o término do processo. Após a finalização, você poderá exibir sua simulação abrindo os arquivos solvatehexanos6.psf e solvatehexanos6.dcd através do VMD.
Você pode melhorar a visualização alterando as configurações gráficas.